Zeyin Yan
Senior Research Fellow
Southern University of Science and Technology
Research Affiliate
Lung Wa CHUNG Group
Home
Research
CV
Code
SocialContact:
Department of Chemistry
Southern University of Science and Technology
1088 Xueyuan Avenue
Shenzhen 518055
P.R. China
Hosted on GitHub Pages — Theme by orderedlist
Published & Forthcoming Papers
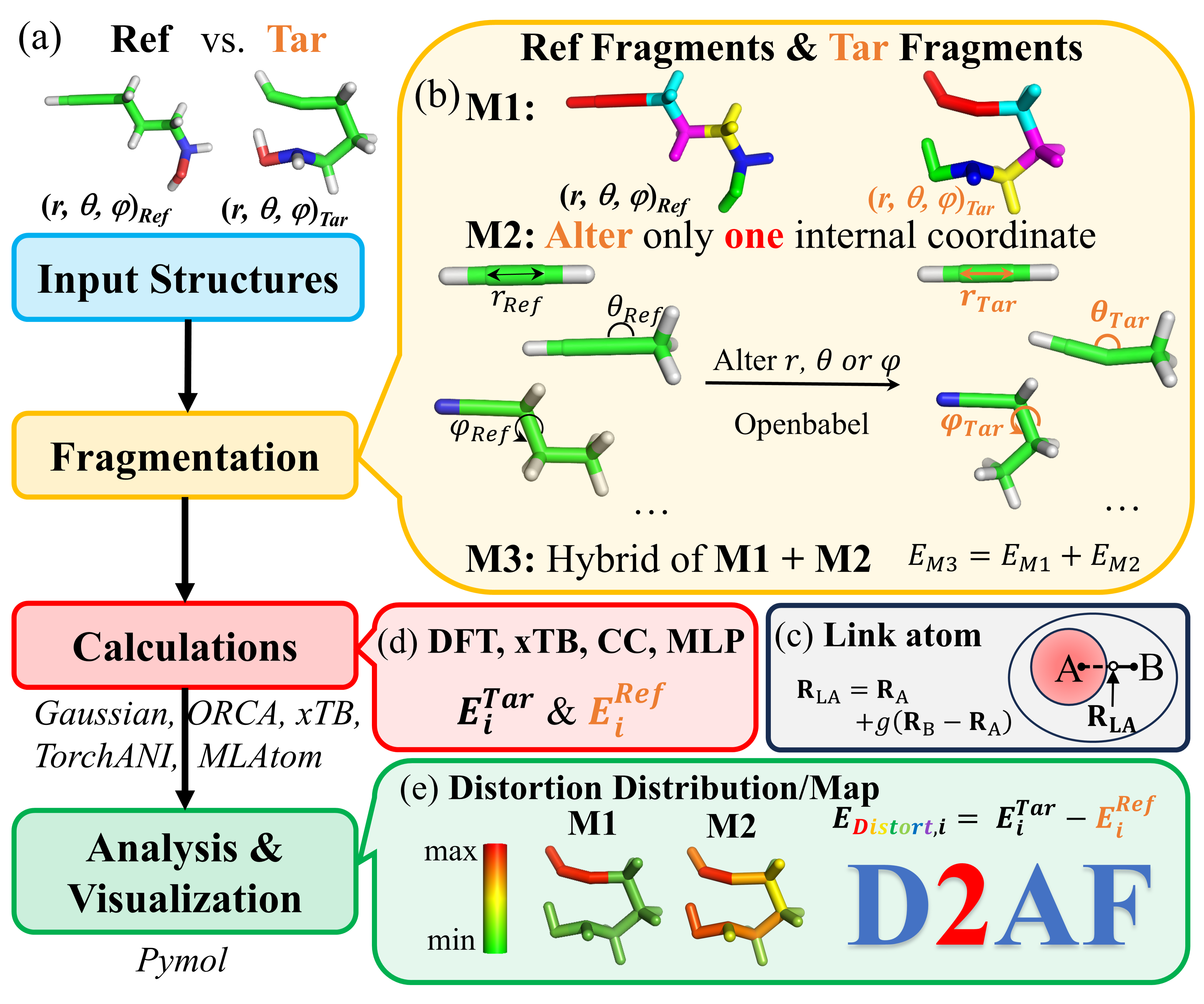
- An efficient and flexible approach for local distortion: distortion distribution analysis enabled by fragmentation
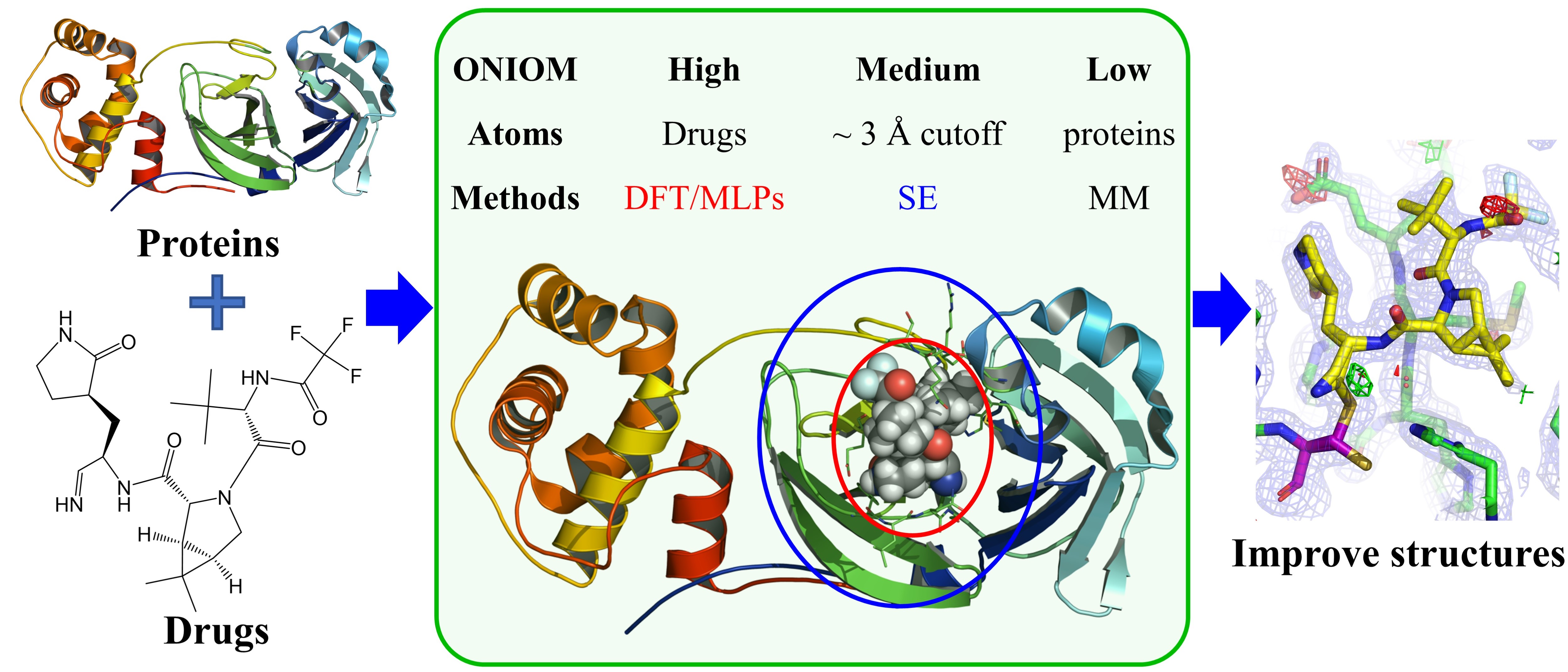
- Accelerating Reliable Multiscale Quantum Refinement of Protein-Drug Systems Enabled by Machine Learning
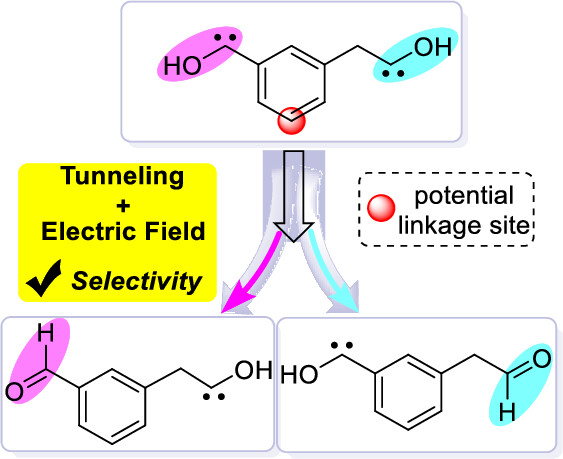
- Quantum Tunneling in Reactions Modulated by External Electric Fields: Reactivity and Selectivity
- Structure-property relationships of photofunctional diiridium(II) complexes with tetracationic charge and an unsupported Ir-Ir bond
- Multiscale Quantum Refinement Approaches for Metalloproteins
- Spin resolved electron density study of YTiO3 in its ferromagnetic phase: signature of orbital ordering
- Joint refinement model for the spin resolved one-electron reduced density matrix of YTiO3 using polarized neutron dffraction and magnetic Compton scattering data
- Development of a joint refinement model for the spin-resolved one-electron reduced density matrix using different data sets
- Spin density in YTiO3: I. Joint refinement of polarized neutron diffraction and magnetic x-ray diffraction data leading to insights into orbital ordering
- Spin density in YTiO3: II. Momentum-space representation of electron spin density supported by position-space results
- When combined X-ray and polarized neutron diffraction data challenge high-level calculations: spin-resolved electron density of an organic radical
- Reactivity of 12-tungstophosphoric acid and its inhibitor potency toward Na+/K+-ATPase: A combined 31P NMR study, ab initio calculations and crystallographic analysis
Authors: Zeyin Yan‡, Y. S. Liao‡, X. Li and Lung Wa Chung*
Chemical Science, 16. 2025
Distortion can play crucial roles in influencing structures and properties, as well as enhancing reactivity or selectivity in many chemical and biological systems. The distortion/interaction model is a popular and powerful method for deciphering the origins of activation energies, in which distortion and interaction energies dictate an activation energy. However, decomposition of local distortion energy at the atomic scale remains less clear and straightforward. Knowing such information should deepen our understanding of reaction processes and improve reaction design. Herein, an efficient, general and flexible fragmentation-based approach was proposed to evaluate local distortion energies for various chemical and biological molecules. Moreover, our distortion analysis is applicable to multiple structures from molecular dynamics (or minimum energy path) as well as can be approximated by different computational chemistry methods. Our systematic analysis shows that our approach not only visualizes distortion distributions within molecules (distortion map) and identifies the key distorted pieces, but also offers deeper understanding and insights into structures, reaction mechanisms and dynamics in various chemical and biological systems. Furthermore, our analysis offers indices of local distortion energy, which can potentially serve as a new descriptor in multi-linear regression or machine learning modelling.
Authors: Zeyin Yan, Dacong Wei, Xin Li, and Lung Wa Chung*
Nature Communications, 2024, 15
Biomacromolecule structures are essential for drug development and biocatalysis. Quantum refinement (QR) methods, which employ reliable quantum mechanics (QM) methods in crystallographic refinement, showed promise in improving the structural quality or even correcting the structure of biomacromolecules. However, vast computational costs and complex QM/MM setups limit QR applications. Here we incorporate robust machine learning potentials (MLPs) in multiscale ONIOM(QM:MM) schemes to describe the core parts (e.g., drugs/inhibitors), replacing the expensive QM method. Additionally, two levels of MLPs are combined for the first time to overcome MLP limitations. Our novel MLPs+ONIOM-based QR methods achieve QM-level accuracy with significantly higher efficiency. Furthermore, our refinements provide the first computational evidence for the existence of bonded and nonbonded forms of the FDA-approved drug nirmatrelvir in one SARS-CoV-2 MPro structure. This study highlights that powerful MLPs accelerate QRs for reliable protein-drug complexes, promote broader QR applications and provide more atomistic insights into drug development.
Authors: Zhifeng Ma†, Zeyin Yan†, Xin Li*, and Lung Wa Chung*
The Journal of Physical Chemistry Letters, 2023, 14
Quantum tunneling and external electric fields (EEFs) can promote some reactions. However, the synergetic effect of an EEF on a tunneling-involving reaction and its temperature-dependence is not very clear. In this study, we extensively investigated how EEFs affect three reactions that involve hydrogen- or (ground- and excited-state) carbon-tunneling using reliable DFT, DLPNO-CCSD(T1), and variational transition-state theory methods. Our study revealed that oriented EEFs can significantly reduce the barrier and corresponding barrier width (and vice versa) through more electrostatic stabilization in transition states. These EEF effects enhance the nontunneling and tunneling-involving rates. Such EEF effects also decrease the crossover temperatures and quantum tunneling contribution, albeit with lower and thinner barriers. Moreover, EEFs can modulate and switch on/off the tunneling-driven 1,2-H migration of hydroxycarbenes under cryogenic conditions. Furthermore, our study predicts for the first time that EEF/tunneling synergy can control the chemo- or site-selectivity of one molecule bearing two similar/same reactive sites.
Authors: Fangrui Zheng, Yuhong Yang, Siye Wu, Shunan Zhao, Yifan Zhu, Huimin Su, Jun-Feng Dai, Zeyin Yan, Lung Wa Chung* & Keith Man-Chung Wong*
COMMUNICATIONS CHEMISTRY, 2022, 159
In contrast to the extensively studied dirhodium(II) complexes and iridium(III) complexes, neutral or dicationic dinuclear iridium(II) complexes with an unsupported ligand are underdeveloped. Here, a series of tetracationic dinuclear iridium(II) complexes, featuring the unsupported Ir(II)–Ir(II) single bond with long bond distances (2.8942(4)–2.9731(4) Å), are synthesized and structurally characterized. Interestingly, compared to the previous unsupported neutral or dicationic diiridium(II) complexes, our DFT and high-level DLPNO-CCSD(T) results found the largest binding energy in these tetracationic complexes even with the long Ir(II)–Ir(II) bond. Our study further reveals that London dispersion interactions enhance the stability cooperatively and significantly to overcome the strong electrostatic repulsion between two half dicationic metal fragments. This class of complexes also exhibit photoluminescence in solution and solid states, which, to our knowledge, represents the first example of this unsupported dinuclear iridium(II) system. In addition, their photoreactivity involving the generation of iridium(II) radical monomer from homolytic cleavage was also explored. The experimental results of photophysical and photochemical behaviours were also correlated with computational studies.
Authors: Zeyin Yan, Xin Li, and Lung Wa Chung*
Journal of Chemical Theory and Computation, 2021, 17, 3783–3796
Biomolecules with metal ion(s) (e.g., metalloproteins) play many important biological roles. However, accurate structural determination of metalloproteins, particularly those containing transition metal ion(s), is challenging due to their complicated electronic structure, complex bonding of metal ions, and high number of conformations in biomolecules. Quantum refinement, which was proposed to combine crystallographic data with computational chemistry methods by several groups, can improve the local structures of some proteins. In this study, a quantum refinement method combining several multiscale computational schemes with experimental (X-ray diffraction) information was developed for metalloproteins. Various quantum refinement approaches using different ONIOM (our own N-layered integrated molecular orbital and molecular mechanics) combinations of quantum mechanics (QM), semiempirical (SE), and molecular mechanics (MM) methods were conducted to assess the performance and reliability on the refined local structure in two metalloproteins. The structures for two (Cu- or Zn-containing) metalloproteins were refined by combining two-layer ONIOM2(QM1/QM2) and ONIOM2(QM/MM) and three-layer ONIOM3(QM1/QM2/MM) schemes with experimental data. The accuracy of the quantum-refined metal binding sites was also examined and compared in these multiscale quantum refinement calculations. ONIOM3(QM/SE/MM) schemes were found to give good results with lower computational costs and were proposed to be a good choice for the multiscale computational scheme for quantum refinement calculations of metal binding site(s) in metalloproteins with high efficiency. Additionally, a two-center ONIOM approach was employed to speed up the quantum refinement calculations for the Zn metalloprotein with two remote active sites/ligands. Moreover, a recent quantum-embedding wavefunction-in-density functional theory (WF-in-DFT) method was also adopted as the high-level method in unprecedented ONIOM2(CCSD-in-B3LYP/MM) and ONIOM3(CCSD-in-B3LYP/SE/MM) calculations, which can be regarded as novel pseudo-three- and pseudo-four-layer ONIOM methods, respectively, to refine the key Zn binding site at the coupled-cluster singles and doubles (CCSD) level. These refined results indicate that multiscale quantum refinement schemes can be used to improve the structural accuracy obtained for local metal binding site(s) in metalloproteins with high efficiency.

Authors: Ariste Bolivard Voufack, Iurii Kibalin, Zeyin Yan, Nicolas Claiser, Saber
Gueddida, Béatrice Gillon, Florence Porcher, Arsen Gukasov, Kunishisa
Sugimoto, Claude Lecomte, Slimane Dahaoui, Jean-Michel Gillet and
Mohamed Souhassoua*
IUCrJ, 2019, 6, 884–894
The present work reports on the charge and spin density modelling of YTiO3 in its ferromagnetic state (TC = 27 K). Accurate polarized neutron diffraction and high-resolution X-ray diffraction (XRD) experiments were carried out on a single crystal at the ORPHé E reactor (LLB) and SPRING8 synchrotron source. The experimental data are modelled by the spin resolved pseudo-atomic multipolar model (Deutsch et al., 2012). The refinement strategy is discussed and the result of this electron density modelling is compared with that from XRD measured at 100 K and with density functional theory calculations. The results show that the spin and charge densities around the Ti atom have lobes directed away from the O atoms, confirming the filling of the t2g orbitals of the Ti atom. The dxy orbital is less populated than dxz and dyz, which is a sign of a partial lift of degeneracy of the t2g orbitals. This study confirms the orbital ordering at low temperature (20 K), which is already present in the paramagnetic state above the ferromagnetic transition (100 K).
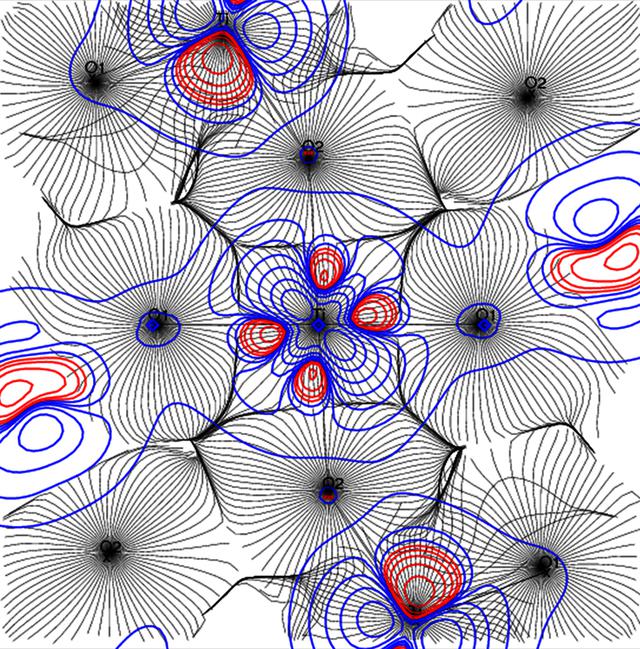
Authors: Saber Gueddida, Zeyin Yan, Iurii Kibalin, Ariste Bolivard Voufack, Nicolas Claiser,
Mohamed Souhassou, Claude Lecomte, Béatrice Gillon, and Jean-Michel Gillet*
The Journal of Chemical Physics., 2018, 148, 164106
In this paper, we propose a simple cluster model with limited basis sets to reproduce the unpaired electron distributions in a YTiO3 ferromagnetic crystal. The spin-resolved one-electron-reduced density matrix is reconstructed simultaneously from theoretical magnetic structure factors and directional magnetic Compton profiles using our joint refinement algorithm. This algorithm is guided by the rescaling of basis functions and the adjustment of the spin population matrix. The resulting spin electron density in both position and momentum spaces from the joint refinement model is in agreement with theoretical and experimental results. Benefits brought from magnetic Compton profiles to the entire spin density matrix are illustrated. We studied the magnetic properties of the YTiO3 crystal along the Ti–O1–Ti bonding.We found that the basis functions are mostly rescaled by means of magnetic Compton profiles, while the molecular occupation numbers are mainly modified by the magnetic structure factors
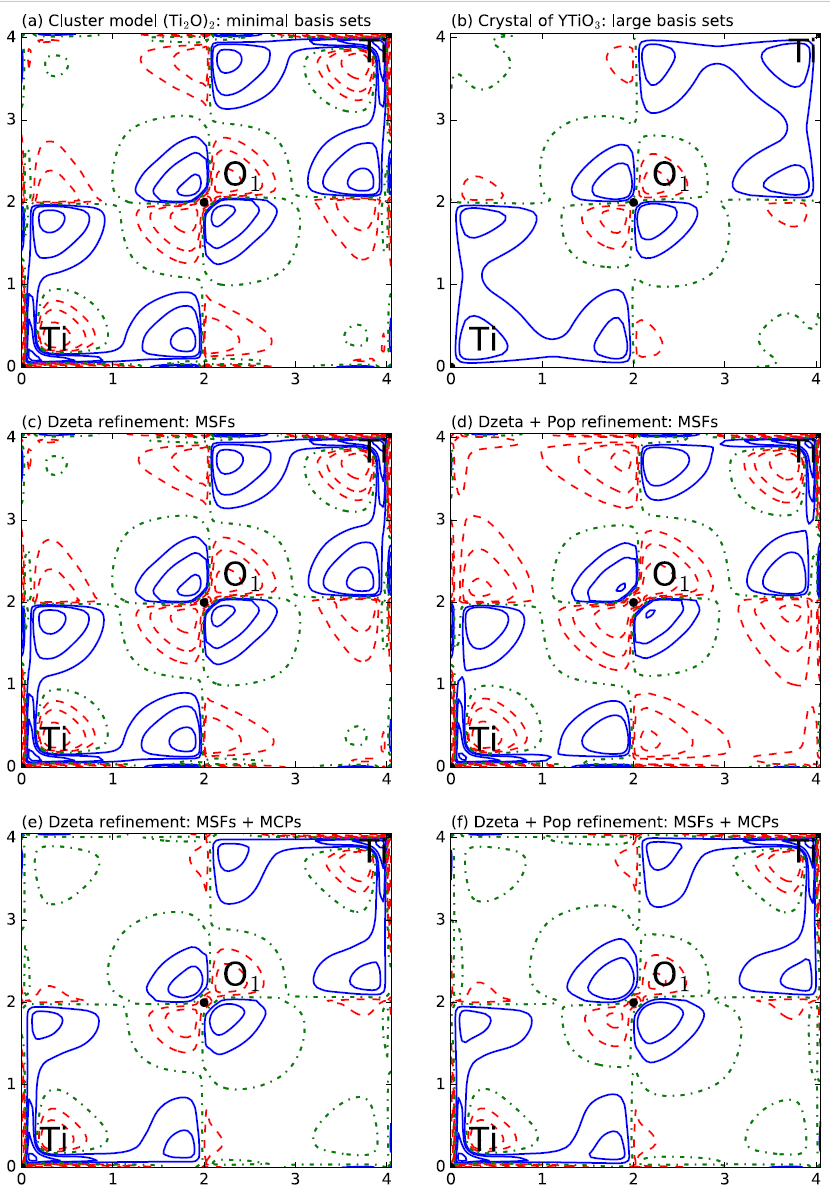
Authors: Saber Gueddida, Zeyin Yan, and Jean-Michel Gillet*
Acta Crystallographica A: Foundation and Advances, 2018, 74, 131-142
The paper describes a joint refinement model of the spin-resolved one-electron reduced density matrix using simultaneously magnetic structure factors and magnetic directional Compton profiles. The model is guided by two strategies: (i) variation of basis functions and (ii) variation of the spin population matrix. The implementation for a finite system is based on an expansion of the natural orbitals on basis sets. To show the potential benefits brought by the joint refinement model, the paper also presents the refinement results using magnetic structure factors only. The joint refinement model provides very satisfactory results reproducing the pseudo-data. In particular, magnetic Compton profiles have a strong effect not only on the off-diagonal elements of the spin-resolved one-electron reduced density matrix but also on its diagonal elements.
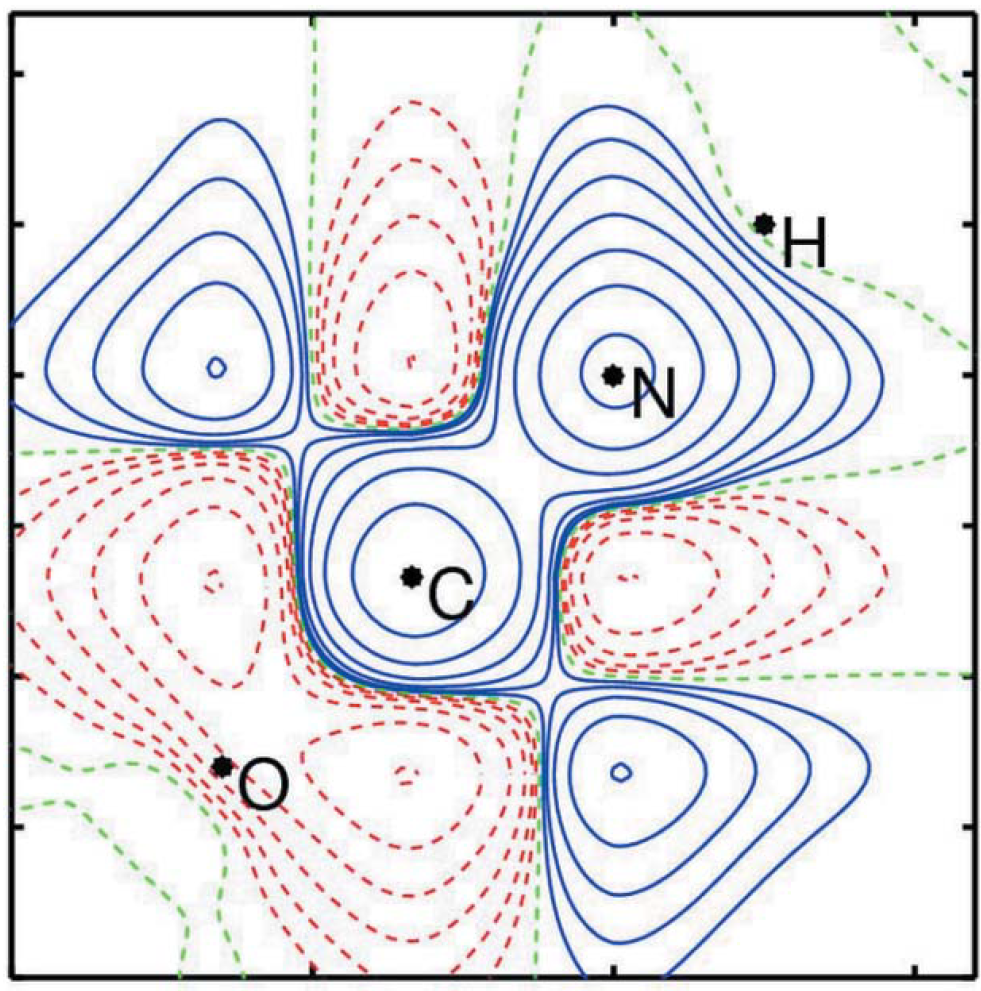
Authors: A. Kibalin, Z. Yan, A. B. Voufack, S. Gueddida, B. Gillon,* A. Gukasov, F. Porcher, A. M. Bataille,
F. Morini, N. Claiser, M. Souhassou, C. Lecomte, J.-M. Gillet, M. Ito, K. Suzuki, H. Sakurai,
Y. Sakurai, C. M. Hoffmann, and X. P. Wang
PHYSICAL REVIEW B, 2017, 96, 054426
Orbital ordering below 30 K was previously observed in the ferromagnetic YTiO3 compound both by polarized neutron diffraction (PND) and x-raymagnetic diffraction (XMD). In this paper we report a procedure for the joint refinement of a unique spin-density model based on both PND and XMDdata. The distribution of the unpaired 3d electron of titanium is clearly seen on the magnetization density reconstructed by the maximum entropy method from the PND data collection at 5 K. The Ti3+ 3d orbital populations obtained by joint model refinement are discussed in terms of the orbital ordering scheme. Small but significant magnetic moments on apical oxygen O1 and yttrium atoms are found. The agreement between experimental and theoretical spin densities obtained using density functional theory is discussed.
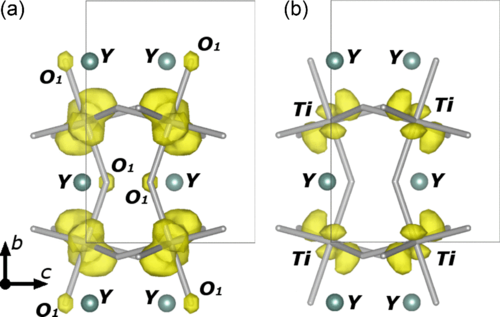
Authors: Z. Yan, I. A. Kibalin, N. Claiser, S. Gueddida, B. Gillon, A. Gukasov, A. B. Voufack, F. Morini, Y. Sakurai,
M. Brancewicz, M. Itou, M. Itoh, N. Tsuji, M. Ito, M. Souhassou, C. Lecomte, P. Cortona, and J.-M. Gillet*
PHYSICAL REVIEW B, 2017, 96, 054427
Unpaired electrons in YTiO3 ferromagnetic crystal (below 30 K) were studied by polarized neutron diffraction (PND) and incoherent x-ray magnetic Compton scattering (MCS). These experiments provide both position and momentum representations of the electrons at the origin of the magnetic behavior, mostly those in the t2g state of Ti atoms. A two-dimensional reconstruction was conducted from experimental and theoretical directional magnetic Compton profiles to obtain the two-dimensional magnetic electron momentum density. A “superposition” method is proposed to examine the coherence between results for position and momentum spaces, respectively. This model-free approach allows a straightforward cross-checking of PND and MCS experiments. An “isolated Ti model” is proposed to emphasize the role played by O1 in the ferromagnetic coupling between Ti and its neighboring atoms.
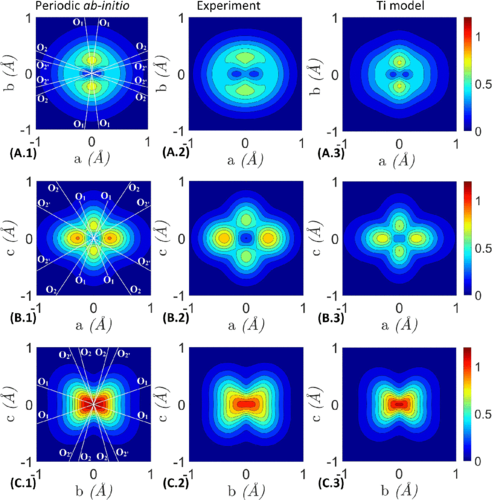
Authors: Ariste Bolivard Voufack, Nicolas Claiser, Claude Lecomte, Sébastien
Pillet, Yves Pontillon, Béatrice Gillon, Zeyin Yan, Jean-Michel Gillet, Marco
Marazzi, Alessandro Genonif, and Mohamed Souhassoua*
Acta Crystallographica Section B: Structural Science Crystal Engineering and Materials, 2017, 73, 544-549
Joint refinement of X-ray and polarized neutron diffraction data has been carried out in order to determine charge and spin density distributions simultaneously in the nitronyl nitroxide (NN) free radical Nit(SMe)Ph. For comparison purposes, density functional theory (DFT) and complete activespace self-consistent field (CASSCF) theoretical calculations were also performed. Experimentally derived charge and spin densities show significant differences between the two NO groups of the NN function that are not observed from DFT theoretical calculations. On the contrary, CASSCF calculations exhibit the same fine details as observed in spin-resolved joint refinement and a clear asymmetry between the two NO groups.
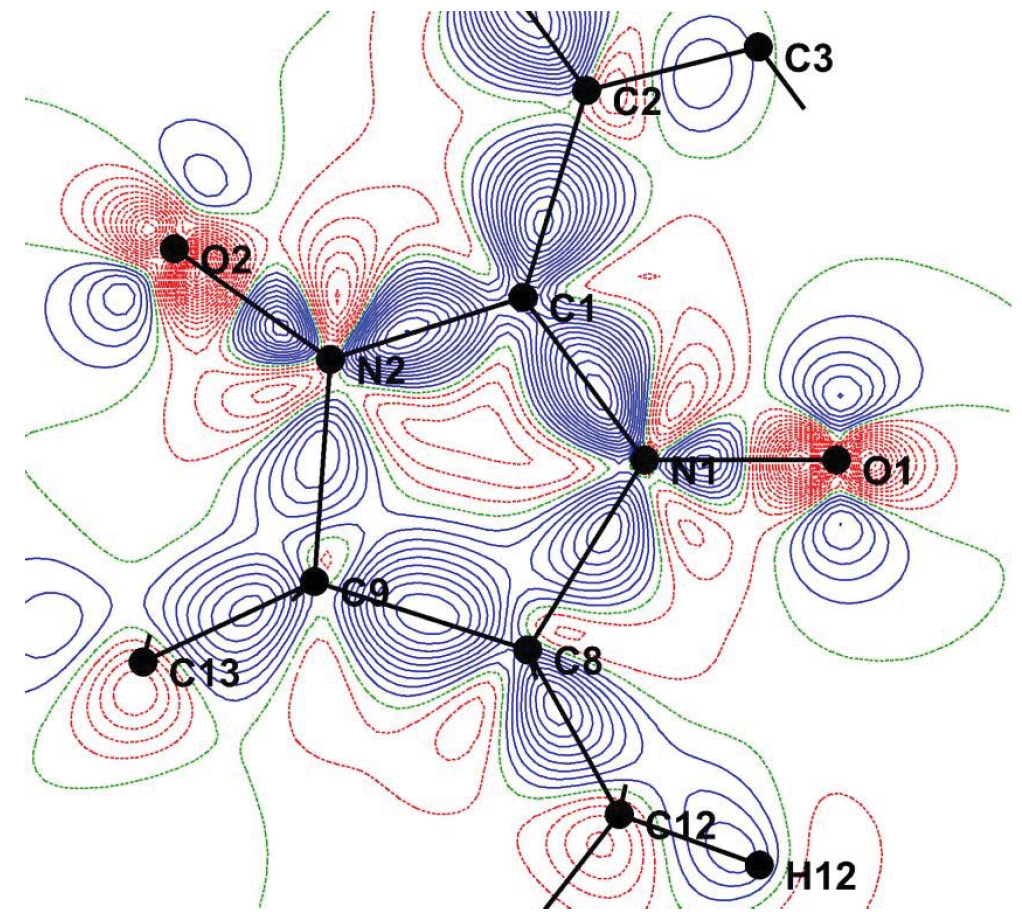
Authors: Nada Bošnjaković-Pavlović⁎, Danica Bajuk-Bogdanović, Joanna Zakrzewskab, Zeyin Yan,
Ivanka Holclajtner-Antunović, Jean-Michel Gillet, Anne Spasojević-de Biré
Acta Crystallographica Section B: Structural Science Crystal Engineering and Materials, 2017, 73, 544-549
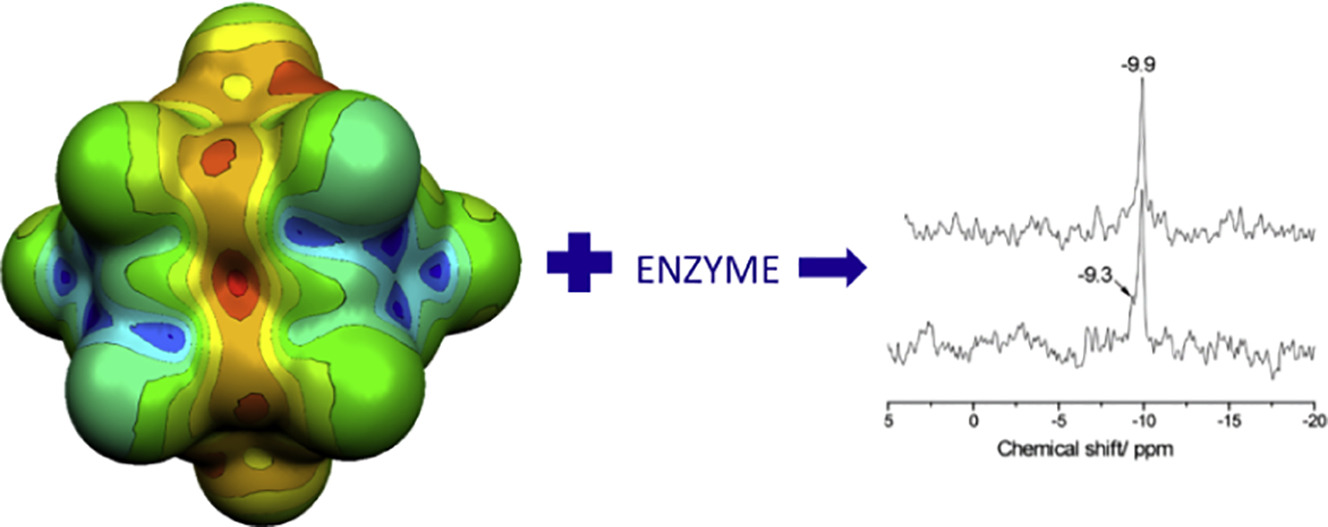
Influence of 12-tungstophosphoric acid (WPA) on conversion of adenosine triphosphate (ATP) to adenosine diphosphate (ADP) in the presence of Na+/K+-ATPase was monitored by 31P NMR spectroscopy. It was shown that WPA exhibits inhibitory effect on Na+/K+-ATPase activity. In order to study WPA reactivity and intermolecular interactions between WPA oxygen atoms and different proton donor types (D =O, N, C), we have considered data for WPA based compounds from the Cambridge Structural Database (CSD), the Crystallographic Open Database (COD) and the Inorganic Crystal Structure Database (ICSD). Binding properties of Keggin's anion in biological systems are illustrated using Protein Data Bank (PDB). This work constitutes the first determination of theoretical Bader charges on polyoxotungstate compound via the Atom In Molecule theory. An analysis of electrostatic potential maps at the molecular surface and charge of WPA, resulting from DFT calculations, suggests that the preferred protonation site corresponds to WPA bridging oxygen. These results enlightened WPA chemical reactivity and its potential biological applications such as the inhibition of the ATPase activity.
Thesis
Reconstruction of momentum density and determination of oneelectron reduced density matrix
Supervisor: Jean-Michel Gillet
Jury: Claude Lecomte, Carlo Gatti,
Enrique Espinosa, Nour-Eddine Ghermani,
Nour-Eddine Ghermani,Béatrice Gillon and Alessandro Erba
Slides: for PhD defense
Works in Progress
1. Quantum refinment on excited-state
2. Protein-Ligand Interaction